Thank you very much for the reply. I'm sure you're sick of answering the same orientation questions. But if you don't mind I have a couple follow-ups...
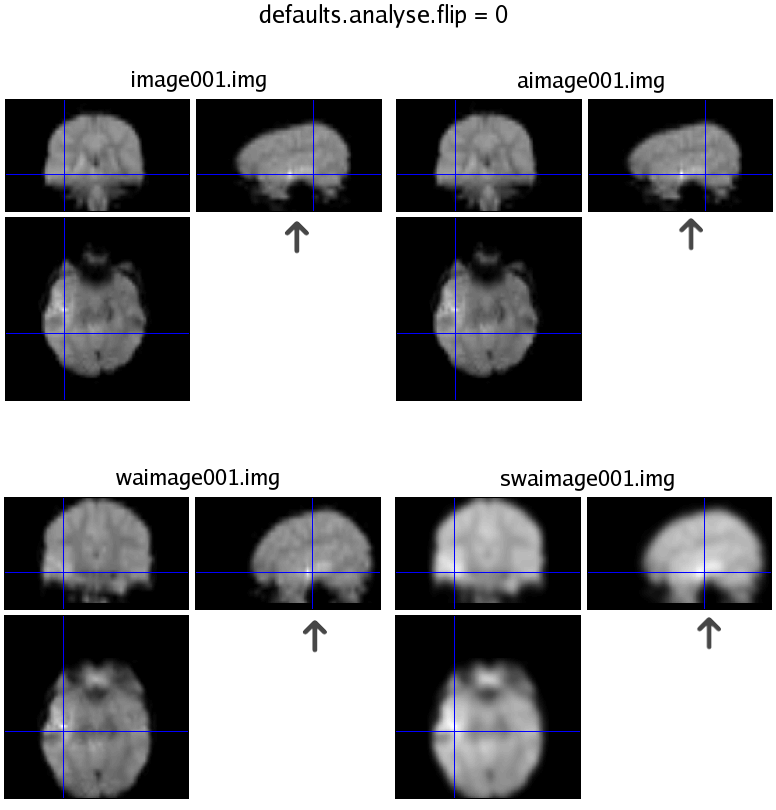
I took a dataset in which I knew for certain which side was which. We scanned a girl w/ a left hemisphere abnormality, and did a verb generation task. Naturally, we would expect the activation to appear in the left hemisphere as well. So, I ran the analysis twice: once with defaults.analyse.flip = 0, and again = 1. The results were mirror images based on what the default was set at. I'm attaching 2 images from check_reg, that are clearly labeled.
As you can see from the images, I have a black arrow pointing at the lesion (it shows up brighter than the rest of the scan. When default = 1, the lesion is flipped to the right after slice-timing and stays that way throughout (the image* files have not gone through any pre-processing; slice-timing is step 1). When the default is 0, the lesion stays on the left side, and when I view the results, the activation is on the left of the image (I overlay results on either a canonical T1, or the co-registered SPGR; I don't use the Slices view). Also, I checked the x-value in mm and in each case the lower x-values were to the left side of the images (both can't be correct, of course).
I understand that the obvious best solution is to convert from dicom to nifti, but for the here and now, I just want a little understanding of how this setting comes into play. On a similar note, when I converted the entire scan (5500 images for an 11 min., TR= 3 session) to a single .nii file, using mri_convert again, SPM5 wouldn't let me slice-time. Is this a bug, or do I need separate files for each volume (i.e. image001 - image219)? I would love to use SPM to convert the DICOM's, but as of now it's been running for over an hour, whereas mri_convert does it in a couple minutes :-/ Any tips on how to speed things up (mri_convert is a pure C program, and, as I understand it, Matlab doesn't handle 5500 file I/O's as quickly).
Thanks again for the help,
Chris
-----Original Message-----
From: John Ashburner [mailto:[log in to unmask]]
Sent: Wed 9/24/2008 7:19 AM
To: Watson, Christopher; [log in to unmask]
Subject: Re: [SPM] More orientation questions...
> Sorry to bring up orientation issues again, but I want to be 100% certain
> that when we view our results, right is right and left is left.
>
> I use Freesurfer's mri_convert to convert the DICOM images (from GE 3T) to
> Analyze format (e.g. image001.{img,hdr,mat}, ..., imageXXX.{img,hdr,mat}).
> Doug Greve from NMR has assured me that mri_convert doesn't change the
> orientation of any of the images. It does output the .mat file containing
> the transformation matrix M.
At this point, I can not be 100% certain either as I don't know enough about
the converter. If there is a way of converting your DICOM to NIfTI format
and if you only use NIfTI compliant software throughout, then there should be
no confusion at all.
>
> Then I use SPM5 to slice-time, realign & reslice, normalise, and smooth. I
> have done analyses with defaults.analyse.flip = 0, and = 1. However, the
> results are exactly the same when I compare them. This is not true of some
> patients we scan, whose data we do NOT normalise, but rather co-register w/
> a structural image (e.g. SPGR). The results were flipped based on the value
> of the default.
>
> This got me thinking:
> 1) Does normalising to the template (e.g. T2.nii) automatically flip the
> images? If so, when I run an analysis, will SPM even bother checking
> defaults.analyse.flip?
Not in SPM5. The flipping only happens with obsolete versions of SPM (SPM99
and earlier).
> 2) Does a value of 0 essentially mean "these images
> are in radiological convention; they need to be flipped to be viewed in
> neurological convention"?
"Radiological convention" is a largely meaningless term when it comes to 3D
images that may be stored sagittally. All it refers to is how an axial image
is displayed - which is not necessarily how it is stored.
> 3) Does the value of defaults.analyse.flip affect
> what I see on the screen? e.g. Will I see something different when I use
> the Results, Display, or Check Reg. functions if I change the value?
This defaults.analyse.flip setting only influences how images are treated when
they are not in NIfTI format (or do not have an SPM5 .mat file). If the
image headers does not encode the orientation, then it is a way of telling
SPM what orientation to assume that the data are in.
The "Slices" view from the results confuses many people as it shows an
original slice from the data - rather than a re-oriented slice as seen in the
orthogonal views. If the left and right appear on the wrong sides, then
think of this as the brain being rotated by 180 degrees. The important thing
is to get the mm coordinates correct. Smaller values for the x coordinate
are towardsthe left of the brain.
>
> The reason this is so important, is that it's easy enough when we have a
> patient w/ a lesion to tell which side is which. But on normal subjects,
> even with the Vitamin E pill on their temple, it doesn't show up on the
> functional images. So it's nearly impossible to tell. We also have DTI
> data, that I manually flipped, so that using SPM's Display function, right
> is right and left is left. However, the FA differences and functional
> differences appear to be in opposite hemispheres... and I want to be sure
> if I'm currently doing something wrong/backwards.
If you can get your data into NIfTI format, and use only NIfTI compliant
tools, then there should be no confusion.
Best regards,
-John
|












{kind=link}
{kind=link}